机器之心报道
又一波研究得出了 LK-99 没有超导性的结论。昨日,北京大学量子材料科学中心(ICQM)郭凯臻、贾爽等人提交到 arXiv 的论文表示,其团队尝试合成的 LK-99 样品不具有超导性。但各路网友似乎对这项研究并不买账,有人认为论文完成度不高,就此宣判 LK-99 的不是超导并不科学。
今日,关于室温超导,国内机构又在 arXiv 上提交了两篇论文。这次下场的有中国科学院物理研究所 / 北京凝聚态物理国家研究中心、中国人民大学、宁波大学、北京师范大学、曲阜师范大学等科研机构和院校。
其中,中国科学院物理研究所 / 北京凝聚态物理国家研究中心所写第一篇论文的结论是 LK-99 的类超导行为是由掺杂 Cu_2S 的一级结构相变引起的;中国科学院物理研究所、人大等机构得出的结论是 P b_10?xCu_x (PO_4)_6O (x = 1) 的基态被确定为半导体相。
我们接下来一一来看两篇论文的具体内容。
中国科学院物理研究所 / 北京凝聚态物理国家研究中心团队
第一篇论文题目为《First order transition in P b_10-xCu_x (PO_4)_6O (0.9<x<1.1) containing Cu_2S》。

论文地址:https://arxiv.org/ftp/arxiv/papers/2308/2308.04353.pdf
研究者发现,报道的 LK-99 含有一定量的 Cu_2S 杂质,它在 400K(约 127 摄氏度)附近经历了从高温 β 相到低温 γ 相的结构相变。为了探究这种类超导转变是 LK-99 固有的还是由 Cu_2S 杂质引起的,研究者研究了 Cu_2S 以及 LK-99、Cu_2S 混合物的输运和磁性质。
结果发现,Cu_2S 的电阻率在 385 K(97 摄氏度)左右下降了 3 到 4 个数量级,接近参考文献中报道的转变温度和电阻行为。此外测量了 LK-99 和 Cu_2S 混合物的电阻率,结果显示在同样的温度下电阻率发生急剧变化,与报道的结果一致,但电阻率并不为 0。
因此,基于对电阻率和磁化强度的测量,研究者认为 LK-99 的类超导行为最有可能是由 Cu_2S 的一级结构相变引起的。
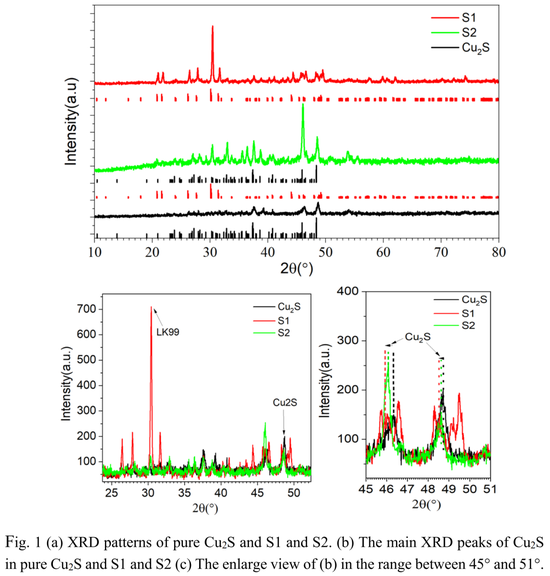
下图 1 (a) 和 (b) 显示了纯 Cu_2S 以及 LK-99、Cu_2S 混合物的 XRD 图谱。Cu_2S 相的 XRD 图谱在具有空间群 P2_1/c 的单斜晶型结构基础上得到了很好索引。LK-99 与数据库索引相匹配,其中含有 Cu_2S 杂质相。并且,反映 Cu_2S 含量的强度比在样品 1(即 S1)中约为 5%,在样品 2(S2)中约为 70%。
图 1 (c) 显示了纯 Cu_2S 中 Cu_2S 以及 S1/S2 分别在 45° 与 51° 之间的主峰。研究者注意到混合物中 Cu_2S 的峰位置与纯 Cu_2S 相比略有位移,这可能是 Cu_2S 中 S 含量的差异导致的。
下图 2 显示了 Cu_2S 在 2 K-400 K 温度范围内的电阻率,分别绘制在线性刻度(图 2(a))和对数刻度(图 2(b))上。
可以看到,在 385 K 左右 Cu_2S 的电阻率显著降低,从 385.6 K 时的 13.7 Ohm cm 降低到 381.5 K 时的 0.006 Ohm cm,下降了 3~4 个数量级。这种电阻率下降与 Lee 等人报道的 LK-99 中的电阻下降类似,后者下降时的温度约为 378K。

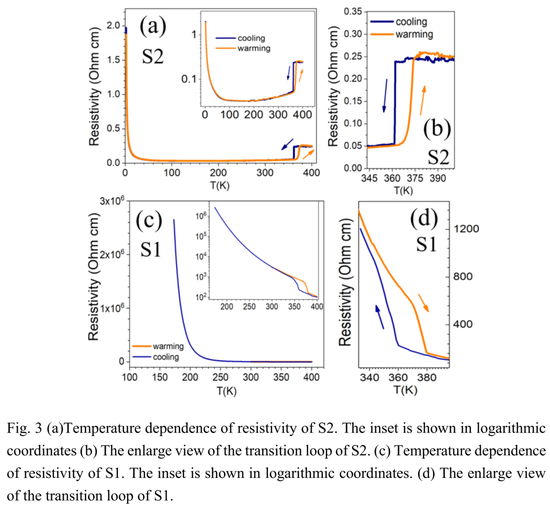
下图 3 (a) 和 (b) 显示样品 2(S2)在 370 K 附近电阻率的急剧下降,并伴有明显的热滞后现象。随着温度降低,电阻率在较宽的高温范围内表现出金属行为 (dρ/dT > 0)。在 100 K 下,电阻率随着温度的降低而增加,表现出类似半导体的行为。这里的电阻率和转变温度的急剧下降与 Lee 等人观察到的类似。
下图 3 (c) 和 (d) 显示了样品 1(S1) 电阻率的温度依赖性。研究者观察到电阻率在 370 K 左右发生跳跃,同样也存在热滞后行为。
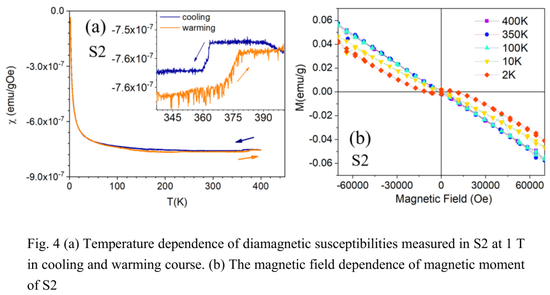
下图 4 (a) 显示了样品 2(S2)在 2 K - 400 K 温度范围内、1 T 磁场中的磁化率。磁化率表现出了抗磁性,并在 375 K 处发生相变。在磁化率数据中再次观察到了超过 10 K 的明显热滞后现象,表明存在一级相变。此外,转变温度范围与 Cu_2S 的结构相变温度密切相关。
下图 4 (b) 显示了磁化强度与磁场的关系,在 2 K - 400 K 的不同温度下,磁化强度范围为 -7 T 到 7 T。这里表现出典型的抗磁行为,与超导体的行为没有相似之处。
对于这项研究和相应结论,华工大佬「洗芝溪」给予了非常正向的评价,认为是目前最专业、最易认可的解释。

完整知乎回答见:https://www.zhihu.com/question/616368545
中国科学院物理研究所、人大、宁波大学等联合团队
第二篇论文题目为《Structural, electronic, magnetic properties of Cu-doped lead-apatite P b_10?xCu_x (PO_4)_6O》。

论文地址:https://arxiv.org/pdf/2308.04344.pdf
最近有关 Cu 掺杂 P bPO 混合物超导性的报道激发了对其物理性质的广泛研究。本文中,研究者探究了该混合物的详细原子和电子结构,这是解释其物理性质(包括可能的超导性)的必要信息。
通过第一性原理的电子结构计算,研究者发现 4f 位点的 P b(而非 6h 位点的 P b)部分被 Cu 原子取代,这对控制费米能级的电子态起到了至关重要的作用。
从结果来看,Cu 原子的 3d 电子轨道出现在费米能附近并表现出强烈自旋极化,从而在掺杂的 Cu 原子周围产生局域矩。因此,他们认为 P b_10?xCu_x (PO_4)_6O (x = 1) 的基态被确定为半导体相,与实验测量结果非常吻合。
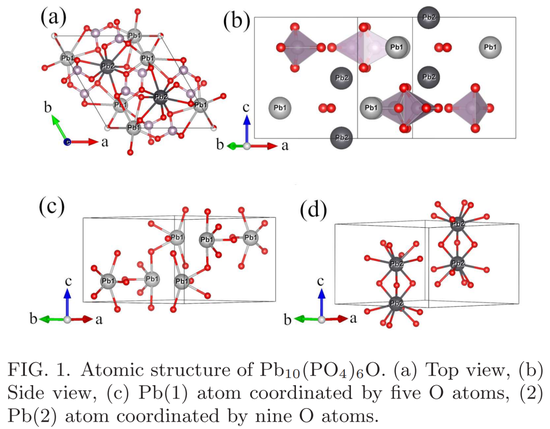
混合物 P b_10 (PO_4)_6O 具有磷灰石型结构,它在空间群 P6_3/m(No.176)中结晶,其中晶体常数 a = 9.8650 ?、c = 7.4306 ?。详细配置如下图 1 所示。
不等价的 Wyckoff 位点形成了两种 P b 原子,分别称为 P b (1) 和 P b (2),它们位于 PO_4 四面体之间的间隙空间。我们可以将 P b (1) 原子和 PO_4 四面体视为 ab 平面上的一层,这些层沿 c 轴方向堆叠,而 P b (2) 原子夹在两层之间。P b (1) 以 5 配位几何结构与五个 O 原子键合。这些 P b-O 的键距为 2.44 - 2.63 ?。P b (2) 以 9 配位几何结构与 9 个 O 原子键合,这些 P b-O 的键距为 2.56 - 2.94 ?。
为了计算方便,研究者在 P b_10?xCu_x (PO_4)_6O 中采用 x = 1,来模拟实验合成的 P b_10?xCu_x (PO_4)_6O (0.9 ≤ x ≤ 1.1) 材料的物理性质。P b 原子有两种,即 6h 位点的 P b (1) 和 4f 位点的 P b (2)。自然的问题是 P b (1) 或 P b (2) 是否被 Cu 原子取代。
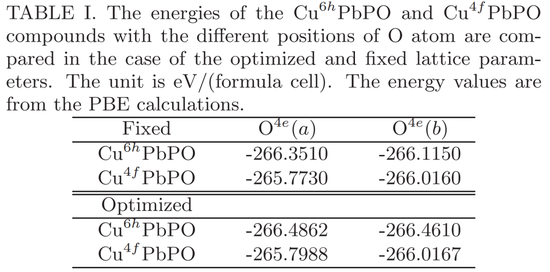
利用测量和优化的晶体参数,他们对 Cu 掺杂铅磷灰石的 P b_9Cu (PO_4)_6O 进行了能量计算。计算出的能量如下表 1 所示,其中 O^4e(a)和 O^4e(b)分别代表 O 原子在 4e 位点的不同位置。
当使用 Cu 原子取代 6h 位点的 P b (1) 而不是 4f 位点的 P b (2) 时,P b_9Cu (PO_4)_6O 具有较低的能量。换言之,掺杂的 Cu 原子应该位于 Cu 掺杂铅磷灰石中的 6h 位点。研究者的结果与 Lee 等人和 Griffin 的结果明显不同。取代 P b (1) 和 P b (2) 原子的 Cu 掺杂铅磷灰石被分别命名为了 Cu^6hP bPO 和 Cu^4fP bPO。
从表中可以看出,O^4e (a) 构型在能量上更有利,这与该 O 原子和掺杂 Cu 原子之间的键合有关。
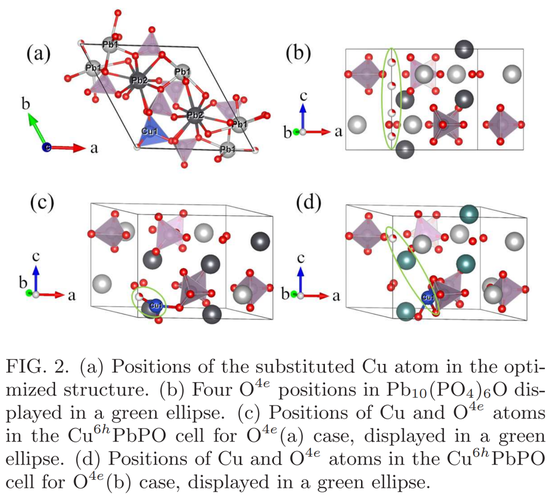
其中,Cu^6hP bPO 的结构如下图 2 (a) 所示。Wyckoff 位点 4e 被 1/4 O 原子占据,与以下四个坐标相关:(0.0 0.0 0.1342)、(0.0 0.0 0.3658)、(0.0 0.0 0.6342) 和 (0.0 0.0 0.8658),如图 2 (b) 所示。由于 Cu 原子的取代,四个 4e 位点变得不等价。这里有必要研究 O^4e 位置对 Cu 掺杂 P bPO 混合物电子性能的影响。
在结构优化开始时,研究者选择坐标(0.0 0.0 0.1342)和(0.0 0.0 0.8658)作为 O^4e 原子的初始位置。优化后,计算中的坐标变为(0.0 0.0 0.1991)和(0.0 0.0 0.7806),分别用 O^4e (a) 和 O^4e (b) 标记,分别如图 2 (c) 和 (d) 所示。
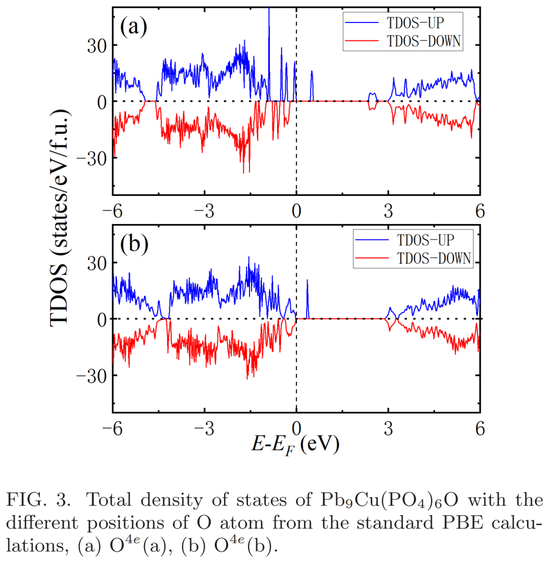
基于能量计算,研究者确定占位 O^4e (a) 的 Cu^6hP bPO 是 P b_10?xCu_x (PO_4)_6O (x = 1) 最可能的结构。总态密度如下图 3(a)所示,表明占位 O^4e(a)的 Cu^6hP bPO 是半导体,能隙约为 0.5 eV。
他们还计算了占位 O^4e (b) 的 Cu6hP bPO 的态密度,如图 3(b)所示。尽管与图 3(a)中的态密度谱有一些明显的差异,但占位 O^4e(b)的 Cu^6hP bPO 也是半导体,能隙约为 0.5 eV。
因此,研究者得出结论,尽管 O^4e 占位不同,但 Cu^6hP bPO 系统仍具有半导体基态。
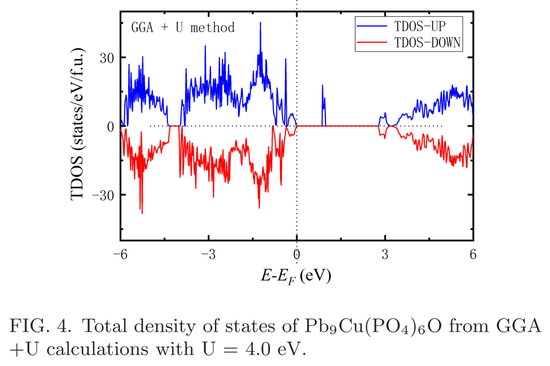
为了进一步确认占位 O^4e (a) 的 Cu^6hP bPO 的半导体基态,研究者采用 GGA+U 方法来检测其电子结构,其中采用的哈伯德 U 值为 4.0 eV,这是之前研究中提出的经验值。总态密度如下图 4 所示,其中能隙约为 0.9 eV,大于 P bE 计算得出的 0.5 eV 值。
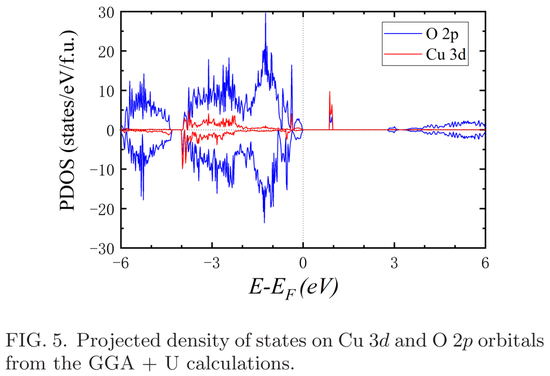
为了展示自旋极化的 Cu 3d 态以及 Cu 3d、O 2p 轨道的杂化,总态密度被投影到原子轨道。下图 5 显示了 Cu 3d 和 O 2p 轨道上的投影态密度,其中 O 2p 轨道的态密度表明 O 2p 和 Cu 3d 轨道之间存在强杂化。另外,Cu 3d 轨道具有明显的自旋极化,这与 Cu 原子周围 1.0 μB 的局域矩有关。
最后为了研究两个掺杂 Cu 原子之间的磁耦合,研究者构建了 1 × 1 × 2 和 2 × 1 × 1 磁性超晶胞,其中两个 Cu 原子的矩以相同或相反的方向排列。
在 1 × 1 × 2 超晶胞中,两个 Cu 原子与 c 轴成一直线,它们的磁相互作用沿 c 方向磁耦合。类似地,2×1×1 超晶胞中的相互作用对应 a 轴方向(或 b 轴)的磁耦合。在研究者的计算中,沿 c 轴方向存在弱铁磁耦合,能量小于 0.1 meV。相较之下,在 ab 平面上,Cu 原子两个矩之间的耦合是反铁磁的,能量小于 1.0 meV。基于此,Cu 掺杂铅磷灰石在基态下的磁性是反铁磁性的。
随着中国科学院物理研究所等科研机构两篇论文的发布,这波室温超导也许已经迎来了「终极答案」。